







蛋白质通常并非以单体形式发挥功能,而是组装成复杂的复合物。由多个相同蛋白质链通过非共价键相互作用形成的复合物被称为同源寡聚体,其空间排列呈现特定对称性,这对于蛋白质的稳定性、折叠和功能至关重要。然而,精确预测蛋白质可能形成的对称结构一直是生物信息学领域的难题。

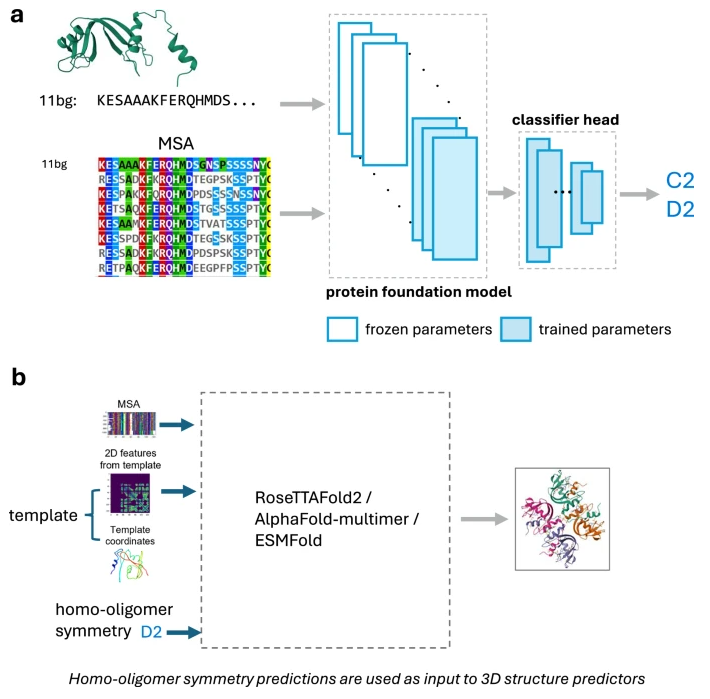
为此,微软、华盛顿大学和麻省理工学院等研究机构合作开发了一种名为Seq2Symm的新方法。该方法通过微调强大的蛋白质语言模型ESM2,实现了每小时约8万个蛋白质的处理能力,其预测准确率显著高于现有方法。这项研究成果已发表在《自然通讯》(Nature Communications)期刊上。


研究突破:Seq2Symm的优势
目前,蛋白质数据库(PDB)中寡聚体状态的注释主要依赖PISA算法,并由研究人员补充。PISA虽然准确性较高,但依赖实验数据。而其他预测方法通常依赖同源模板搜索或基于对接的模拟,计算成本高且受限于高质量多序列比对(MSA)数据的可用性。
Seq2Symm则不同,它仅需单条蛋白质序列作为输入,就能高效准确地预测同源寡聚体的对称性。其核心在于对ESM2模型的巧妙微调,并采用了一种高效的架构,包括一个经过优化的分类器头部模块。Seq2Symm的预测结果可以直接用于指导AlphaFold2-multimer等工具,生成原子精度的同源寡聚体结构模型。

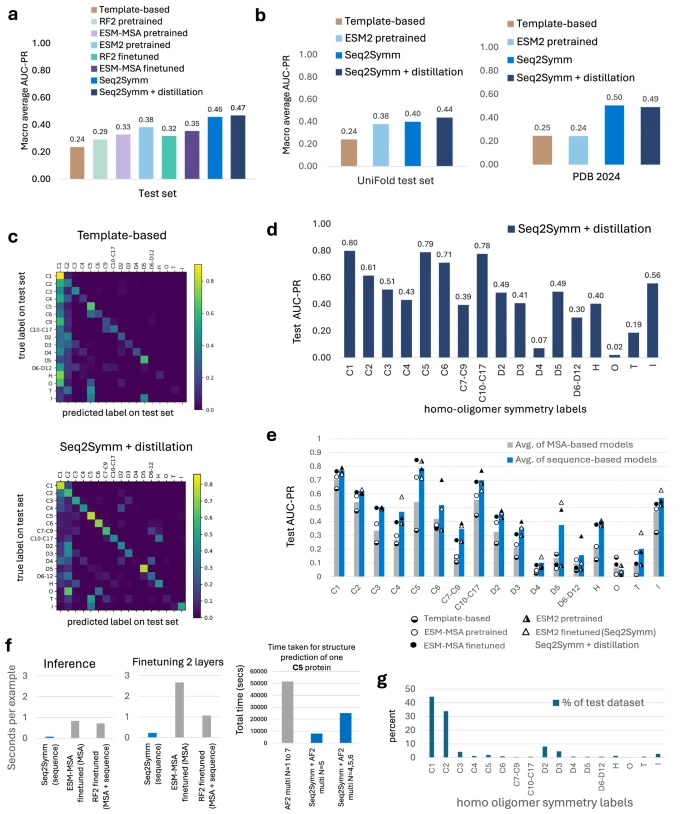
性能评估与应用
Seq2Symm在多个独立测试集上均显著优于基于模板的搜索方法,尤其在预测复杂对称性方面表现突出。令人意外的是,基于单序列的Seq2Symm反而优于基于MSA的模型,这可能是因为同一蛋白质家族成员可能具有不同的同源寡聚体对称性,MSA引入的共进化信号反而可能造成干扰。

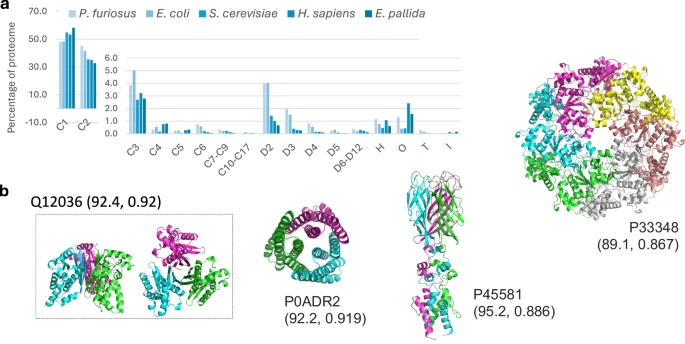
Seq2Symm的高效性使其能够在蛋白组规模上进行应用。研究团队将其应用于多个生物体的蛋白组,揭示了不同生物体中同源寡聚体对称性的分布规律。
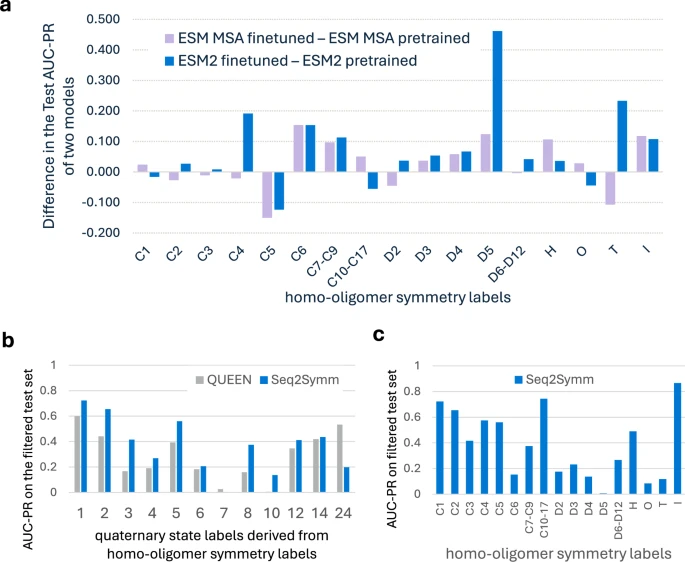




未来展望
尽管Seq2Symm取得了显著进展,但仍有改进空间,例如提高在概率为0.5-0.7的区域内的预测准确性,并减少数据集中标签噪声的影响。未来的研究方向可能包括调整损失函数、整合蛋白质单链结构信息、直接预测粗粒度对称性以及同时预测对称类型和四级结构。
即便如此,Seq2Symm已经为蛋白质结构预测和蛋白组学研究提供了强大的工具,将在未来的生物学研究中发挥重要作用。 论文链接:https://www.php.cn/link/bb6d2babd7797d94d8f4a8600bc9b44e 代码链接:https://www.php.cn/link/bb6d2babd7797d94d8f4a8600bc9b44e
以上就是每小时处理80,000个蛋白质,大卫·贝克、微软等发布Seq2Symm,实现蛋白质对称性精准预测的详细内容,更多请关注【创想鸟】其它相关文章!
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至253000106@qq.com举报,一经查实,本站将立刻删除。
发布者:PHP中文网,转转请注明出处:https://www.chuangxiangniao.com/p/3048512.html

